
GABA synthesis and release from glia:
Our group has contributed to the field of gliotransmission (release and function of transmitters from glia) with seminal publications on the channel-mediated GABA and glutamate release from astrocytes. We demonstrated, for the first time, the channel-mediated release mechanism of the major inhibitory transmitter GABA from astrocytes in cerebellum involving Best1 channel and further demonstrated that the source of tonic inhibition in cerebellum is astrocytic GABA. We subsequently identified the biosynthetic pathway for GABA in astrocyte to be the putrescine degradation pathway leading to GABA production, in which the monoamine oxidase B (MAO-B) is the key enzyme for GABA production.
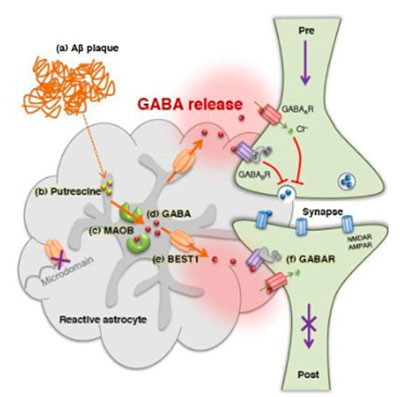
This series of studies implicates that astrocytes by tonically releasing the major inhibitory transmitter GABA, can exert strong inhibitory drive to the brain activity, raising a profound possibility that astrocytes can directly participate in cognitive processes via astrocytic GABA. Utilizing these newly identified molecular targets, Best1 and MAO-B, we further demonstrated that tonic GABA inhibition in cerebellum is critical for motor coordination.
In parallel, we have revealed the pathological role of astrocytic GABA, especially in Alzheimer’s disease (AD) and Parkinson’s disease (PD). We provide compelling pieces of evidence that hippocampal or SNpc astrocytes aberrantly produce GABA via MAO-B and release through Best1 channels in AD or PD, respectively. The aberrant GABA from astrocytes inhibits neighboring neuronal activity to impair memory in AD or to cause motor deficit in PD. We further propose that the astrocytic GABA could be an optimal biomarker, diagnostic tool, and therapeutic target for AD and PD. We continue to investigate the role of aberrant GABA from astrocytes in white matter stroke, recovery after spinal cord injury, obesity, and epilepsy.
The molecular mechanism of glutamate and d-serine release from glia:
In addition to GABA, we have investigated the molecular mechanism of glutamate release from astrocytes because glutamate has been known to be released from astrocytes but the release mechanism has been controversial. Glutamate is the major excitatory transmitter that enhances neuronal excitability.
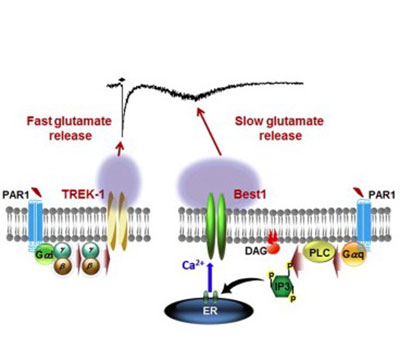
Through an intense investigation, we have clearly demonstrated that glutamate is released in two models: fast mode through TREK-1 containing K2P channel and in slow mode through Best1 channel in hippocampal astrocytes. We have subsequently reported that Best1-mediated glutamate release contributes to receptor mediated synaptic plasticity in hippocampus upon activation of PAR1 (a Gq-coupled GPCR). Through this series of studies, we have demonstrated that astrocytic glutamate and GABA are the key modulator for excitation-inhibition balance (E/I balance) in the brain, which is mainly dependent on the levels of glutamate and GABA. In addition to glutamate, we found that Best1 is also capable of releasing d-serine, which can act as a co-agonist of NMDA receptors to participate in synaptic plasticity. We are actively investigating the possibility of astrocytes as the potential therapeutic target for various brain disorders such as schizophrenia, autism, ADHD, epilepsy, depression, and etc, which are known to be caused by impaired E/I balance.
Astrocytic volume transient and brain plasticity:
We have also contributed to the field of glioscience by identifying and characterizing several astrocytic ion channels. We identified that astrocytic two-pore potassium channel K2P, which is responsible for passive conductance with a subunit composition of a heterodimer of TWIK-1 and TREK-1. We further proposed the heterodimer of TWIK-1 and TREK-1 as the potential therapeutic target of epilepsy, depression, and anxiety disorders caused by dysregulation of potassium ion concentration. Next, we found the identity of astrocytic volume-regulated anion channel (VRAC), which is tweety-homolog (Ttyh).
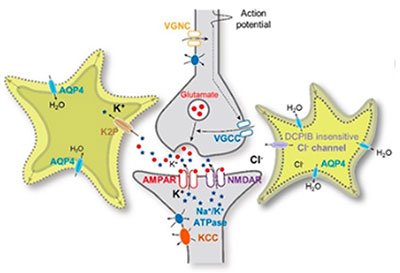
So far the identity of VRAC has been proposed to be LRRC8. However, we firstly demonstrated that the pore-forming subunit of VRAC in astrocytes is Ttyh, but not LRRC8. More importantly, we have demonstrated for the first time that astrocytic volume change through aquaporin-4 water channel is critical for synaptic plasticity and established the novel concept of volume plasticity in both mouse and human. We have demonstrated that this astrocytic volume change is critical for spatial memory in mouse and language-associated learning and memory in human. This ground-breaking finding and novel concept of astrocytic volume will be the basis for a paradigm shift in brain plasticity from the traditional view of neuroplasticity to the new concept of glioplasticity and memory.
Reactive gliosis and neurodegeneration:
During the course of delineating the molecular and cellular mechanisms of gliotransmission, we have encountered the important concept of reactive gliosis, which is often referred to as the basis for neuroinflammation.
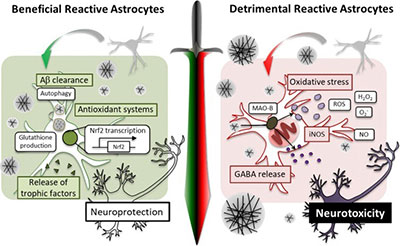
Although pathologic contributions of reactive astrocytes have been implicated in AD and other neurodegenerative diseases, their in vivo functions have remained elusive due to the lack of an appropriate experimental model. We have recently developed an astrocyte-specific toxin receptor model, and found that the astrocytes, which have an active autophagy system, not only remained alive but selectively became reactive. Our study identifies the severe reactive astrocytes as a key determinant of neurodegeneration in AD. We anticipate that this novel mechanistic insights will be very useful for developing drug candidates for AD and other neurodegenerative diseases, such as PD, traumatic brain injury, stroke, and etc.